Design Controls for Medical Devices: 21 CFR 820.30 in Practice
By Jason Brumwell

Design controls for medical devices are the single regulatory mechanism that connects user needs to a verified, validated product. 21 CFR 820.30 defines nine elements that every Class II and Class III device must follow. For software teams, these nine elements map to activities you are already doing: writing requirements, reviewing code, running tests, deploying to production. The engineering is usually fine. What trips teams up is the documentation, traceability, and evidence that proves you did it.
For ContourCompanion, a Class II AI SaMD generating radiation therapy contours from DICOM CT data, design controls ran from the first user need ("clinician reviews AI-generated contour before treatment planning") through to production deployment on AWS. The design history file compiled everything: requirements, architecture decisions, risk controls, test results, deployment records. When the FDA reviewer opened the traceability matrix, every row was complete: requirement to test to risk control, nothing missing.
The Nine Elements of 21 CFR 820.30
The FDA's Design Control Guidance for Medical Device Manufacturers provides the canonical interpretation. The guidance includes a waterfall diagram (originally sourced from Health Canada) that teams misread as a mandated process. It is not. The diagram shows the relationship between elements, not the order you must execute them. AAMI TIR45:2012 explicitly supports agile practices in medical device software development, and the FDA has recognized it as a consensus standard since 2013.

Here is how each element maps to SaMD development:
Design and Development Planning (820.30(b))
The design plan identifies the software safety classification (IEC 62304 Class A/B/C), the documentation level (Basic or Enhanced per the 2023 FDA software guidance), and the regulatory pathway. For ContourCompanion, the plan specified Class C safety classification, Enhanced documentation level, and the 510(k) pathway. The plan updated as the design evolved, which the regulation explicitly requires.
Design Input (820.30(c))
Design inputs are the requirements. For SaMD, these translate to a Software Requirements Specification (SRS) covering functional requirements, performance requirements, interface specifications, cybersecurity requirements, and usability requirements.
The gap that causes FDA deficiencies: teams write design inputs as vague user stories. A user need is not a design input. The user need "clinician reviews AI-generated contour before treatment planning" must decompose into measurable design inputs. Here is what that decomposition looks like for an autocontouring SaMD:
| User Need | Design Input (Measurable) |
|---|---|
| Clinician reviews contour before use | System shall display contours overlaid on source CT within a defined latency threshold |
| Contours distinguish anatomical structures | Each OAR shall render with unique color coding per the RT Structure Set specification |
| Clinician can reject inaccurate contours | System shall require clinician review and approval before export to treatment planning |
| System handles multiple imaging modalities | System shall accept DICOM CT, MRI, and PET/CT inputs conforming to DICOM PS3.3 IOD specifications |
Design inputs must be specific enough that design verification can objectively confirm they were met. "Display contours quickly" fails. "Display contours within [X] seconds" passes. If verification cannot produce a binary pass/fail result, the design input is not written correctly.
Risk-derived requirements also count as design inputs. Every risk control identified in the ISO 14971 risk management file becomes a design input. For ContourCompanion, the risk analysis identified dozens of hazards. Each hazard with an unacceptable residual risk produced at least one risk control, and each risk control produced a design input requirement. A hazard like "algorithm produces inaccurate contour for a critical OAR" can generate multiple design inputs: an accuracy threshold, a mandatory clinician review step, and visual indicators that flag structures needing closer inspection.
Design Output (820.30(d))
Design outputs are the results of the design process: software architecture, detailed design documents, source code, and executable code. The regulation requires that outputs are documented in terms that allow evaluation against design inputs. The key phrase is "traceable to design inputs." Every output must link back to the input it satisfies.
For SaMD, the architecture document is a critical design output. The HIPAA-compliant AWS architecture for ContourCompanion was a design output that satisfied multiple design inputs: PHI encryption requirements, availability requirements, performance requirements for inference latency.
Design Review (820.30(e))
Design reviews are formal, documented examinations of design results. They must include individuals not directly responsible for the design stage being reviewed. In agile terms: sprint reviews, architecture reviews, and risk reviews all qualify, provided they are documented and include cross-functional participants.
The documentation requirement is where agile teams get 483 observations. A sprint review where the product owner approves the demo is not a design review unless there is a record. FDA investigators look for: who attended (with roles), what design outputs were reviewed (with document references), what issues were identified, what corrective actions were assigned, and evidence of follow-through.
For ContourCompanion, design review records captured the sprint number, the specific design outputs under review (linked by document ID), attendees including at least one person not on the development team, identified issues with severity classification, and action items with owners and resolution dates. A review that found no issues still produced a record stating so. The absence of a record is a finding. The presence of a record stating "no issues" is not.
Design Verification (820.30(f))
Design verification answers: "Did we build it right?" It confirms that design outputs meet design input requirements through objective evidence. For software, this maps to unit tests, integration tests, system tests, static analysis, and code reviews.
The FDA expects a clear distinction between verification and validation in the premarket submission. A common deficiency is conflating the two: running unit tests is verification, not validation. Unit tests confirm the code meets its specification. They do not confirm the device meets user needs.
For ContourCompanion, verification included automated test suites running in CI on every pull request: unit tests for algorithm components, integration tests for the DICOM pipeline, system tests for the full ingestion-inference-delivery workflow. Static analysis flagged code quality issues. Every test traced to a specific design input requirement.
Design Validation (820.30(g))
Design validation answers: "Did we build the right thing?" It demonstrates that the device meets user needs and intended uses under actual or simulated use conditions. The regulation text states explicitly: "Design validation shall include software validation and risk analysis, where appropriate."
Validation must happen on production-equivalent devices or systems. For a cloud-deployed SaMD, that means testing against the actual AWS infrastructure, not a local development environment. ContourCompanion's validation included clinical evaluation using standard accuracy metrics (Dice Similarity Coefficients, Hausdorff distances) against expert-drawn contours, usability testing with radiation oncologists on the production interface, and performance validation under production load conditions.
Design Transfer (820.30(h))
Design transfer ensures the final release matches the verified and validated version. For traditional hardware devices, this is manufacturing process validation. For SaMD, design transfer is the deployment process: infrastructure as code, container image builds, release pipelines, and configuration management.
ContourCompanion's design transfer was the CDK deployment pipeline. The IaC template was the production specification. The IQ/OQ/PQ qualification verified that the deployed infrastructure matched the specification. The container image was built from a tagged commit, scanned, and deployed through the same pipeline every time. No manual steps, no console modifications, no gap between the validated design and the production system.
Design Changes (820.30(i))
Every software change, patch, or update must follow change control procedures. The FDA notes that most software-related device recalls involve devices running revised software. Significant changes may trigger a new 510(k).
Pull requests with mandatory review, linked to requirements and test results, are a natural change control mechanism for software teams. The key addition for regulated development: an impact assessment. Before approving a change, evaluate whether it affects previously validated elements. If it does, re-verification and potentially re-validation are required.
Design History File (820.30(j))
The design history file (DHF) compiles all records demonstrating the design was developed per the approved plan. It contains or references: design plans, inputs, outputs, review records, verification evidence, validation evidence, risk management records, design transfer evidence, and change records.
The DHF is not a document you write at the end. It accumulates throughout development. Teams that try to reconstruct a DHF before submission spend months recreating evidence that should have been captured as the work happened. For ContourCompanion, the DHF was assembled incrementally: every sprint produced artifacts that fed into it. By the time the 510(k) submission was assembled, the DHF was already complete.
Design Controls and the Traceability Matrix
The traceability matrix is the artifact that ties design controls together. It demonstrates bidirectional traceability across the full chain:
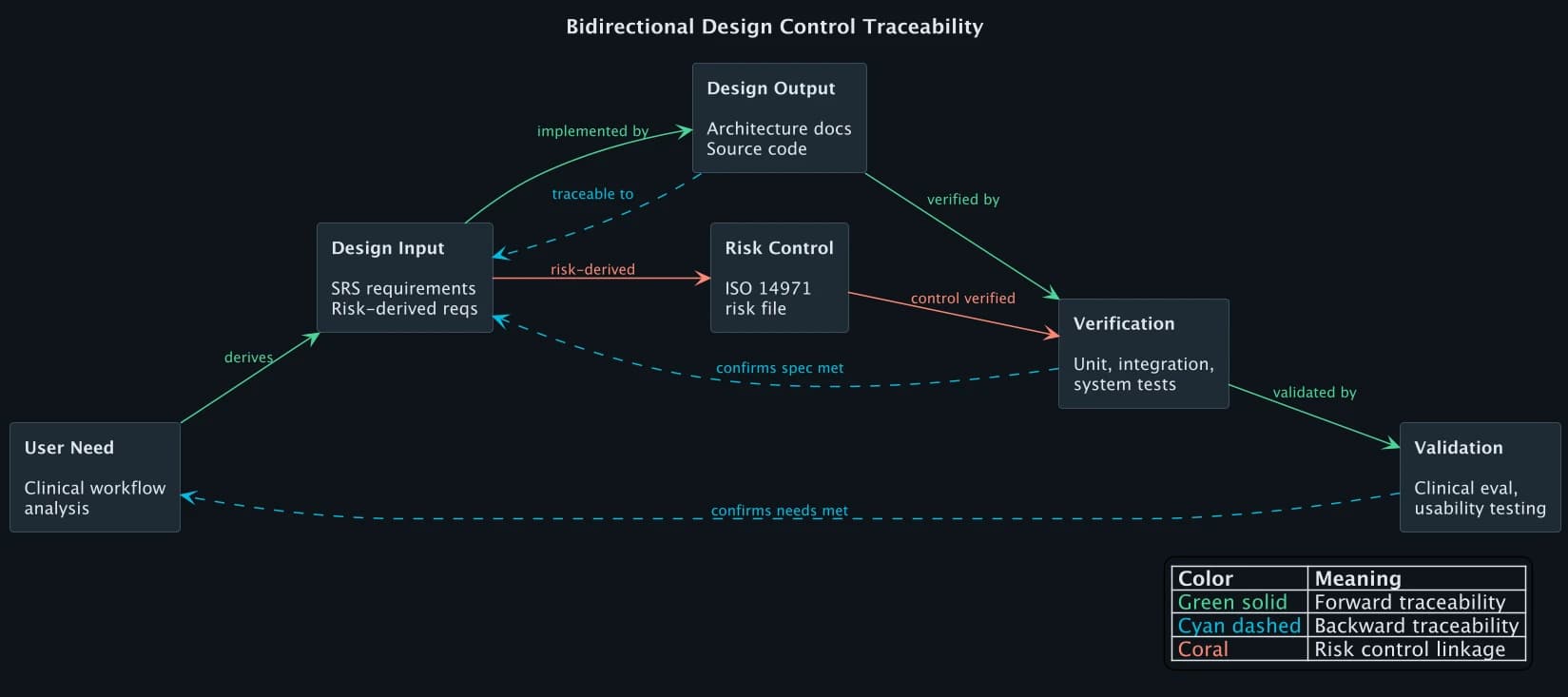
Forward traceability ensures every requirement is implemented and tested. Backward traceability ensures every design element, test, and output has a justified origin. If a test exists without a corresponding requirement, it is orphaned. If a requirement exists without a verification activity, the traceability matrix has a gap. FDA reviewers scan for both.
Here are four rows from ContourCompanion's traceability matrix, showing how different types of requirements (functional, performance, security, risk-derived) each follow the same chain:
| User Need | Design Input | Design Output | Verification | Validation |
|---|---|---|---|---|
| Clinician reviews contour before use | Display contours on CT within defined latency threshold, per-structure color coding | Delivery service, RT Structure Set formatter | Integration test: delivery within threshold (pass) | Usability test: oncologists complete review workflow |
| PHI protected in transit and at rest | All DICOM data encrypted AES-256 at rest, TLS 1.2+ in transit | KMS encryption config, ALB TLS policy | CDK assertion: encryption enabled on all PHI stores (pass) | Pen test: no cleartext PHI in transit (pass) |
| System available during clinical hours | Uptime SLA with Multi-AZ deployment | RDS Multi-AZ cluster, ECS across AZs | IQ script: Multi-AZ active (pass) | Production uptime monitoring over defined period |
| Inaccurate contour must not reach treatment plan | Mandatory clinician review before export to treatment planning system | Review gate in clinical workflow | Integration test: export blocked without review (pass) | Clinical eval: accuracy metrics against expert-drawn contours |
The matrix had columns for user needs, system requirements, software requirements, design outputs, verification tests, validation activities, and risk controls from the ISO 14971 risk management file. The FDA reviewer could pick any requirement and trace it forward to a test result, or pick any test and trace it backward to the user need that justified it.
Where Design Controls Go Wrong
FDA 483 observations and submission deficiencies related to design controls follow predictable patterns. These are the issues that delay clearances and trigger inspection findings:
Conflating verification with validation. Running your automated test suite is verification. It proves the software meets its specifications. It does not prove the device meets user needs. FDA reviewers flag this when the submission presents unit test results as validation evidence. Validation requires testing under actual or simulated use conditions, by representative users, on production-equivalent systems.
Retroactive DHF construction. Teams that treat the DHF as a submission deliverable rather than a living record end up reconstructing months of design history from memory, commit logs, and Slack messages. The evidence is either missing or lacks the contemporaneous timestamps and approvals that FDA expects. The DHF must accumulate as development progresses.
Incomplete traceability. A requirement without a verification test is a gap. A test without a traced requirement is an orphan. Both are findings. The traceability matrix must be bidirectional and complete. Partial traceability (forward only, or only for high-risk requirements) does not satisfy the regulation.
Design reviews without independent reviewers. 820.30(e) requires that reviews include individuals not directly responsible for the design stage being reviewed. A code review by the developer's sprint teammate does not meet this requirement. The review must include at least one person outside the immediate development team, and the record must identify participants by name and role.
Design changes without impact assessment. Changing a software module that affects a previously validated function requires re-verification and potentially re-validation. Teams that treat every pull request the same, regardless of impact on validated elements, accumulate risk. The change control process must include a documented assessment of whether the change affects previously verified or validated design outputs.
QMSR: What Changed in 2026
The FDA's Quality Management System Regulation (QMSR) became effective February 2, 2026, replacing the prior Quality System Regulation (QSR). The QMSR incorporates ISO 13485:2016 by reference rather than maintaining a separate FDA-specific set of requirements.
For design controls, the practical impact is moderate. ISO 13485 Clause 7.3 covers design and development with the same fundamental elements: planning, inputs, outputs, reviews, verification, validation, transfer, and changes. The DHF model persists with harmonized terminology. Teams already compliant with ISO 13485 see reduced burden. Teams that built their QMS around 820.30 specifically need to map their existing procedures to ISO 13485 Clause 7.3, but the substance is the same.
The larger significance: international alignment. A single QMS built to ISO 13485 now satisfies both FDA requirements (via QMSR) and EU MDR requirements, eliminating the need for parallel quality systems. For teams building SaMD for global markets, this is a meaningful reduction in regulatory overhead.
Frequently Asked Questions
What are design controls for medical devices?
Design controls are the set of practices and procedures under 21 CFR 820.30 that ensure a medical device meets specified requirements and user needs throughout development. They consist of nine elements: planning, design input, design output, design review, design verification, design validation, design transfer, design changes, and the design history file (DHF). Design controls are required for all Class II and Class III devices, and for Class I devices containing software.
What is a design history file (DHF)?
A design history file is the compilation of all records that demonstrate a medical device was developed following the approved design control process. It contains or references design plans, requirements (design inputs), architecture and specifications (design outputs), review records, test results (verification and validation evidence), risk management records, design transfer records, and change control documentation. The DHF must be maintained throughout development, not assembled retroactively before submission.
What is the difference between design verification and design validation?
Design verification confirms that design outputs meet design input requirements ("did we build it right?") through objective evidence such as testing, inspection, and analysis. Design validation demonstrates that the finished device meets user needs and intended uses under actual or simulated use conditions ("did we build the right thing?"). Verification tests against specifications. Validation tests against clinical and user requirements. Both are required under 21 CFR 820.30 and must be clearly distinguished in FDA premarket submissions.
References
- 21 CFR 820.30 — Design Controls
- FDA Design Control Guidance for Medical Device Manufacturers
- ISO 13485:2016 — Medical Device Quality Management Systems
- AAMI TIR45:2012 — Agile in Medical Device Software
- FDA QMSR Final Rule (2024)
- FDA Content of Premarket Submissions for Device Software Functions (2023)
- Complizen: FDA Design Controls Complete Guide
- Greenlight Guru: Design Controls for 510(k)
SaMD Engineering
Part 8 of 11
- 1.What Is SaMD? Software as a Medical Device Explained
- 2.FDA Pathways for SaMD: 510(k) vs De Novo vs PMA
- 3.IEC 62304 in Practice: Medical Device Software Without the Waterfall
- 4.ISO 14971 Risk Management for SaMD: What FDA Reviewers Read
- 5.HIPAA Compliant AWS Architecture for Medical Device Software
- 6.Infrastructure as Code for Medical Devices: IQ OQ PQ with AWS CDK
- 7.Medical Device Cybersecurity: FDA Guidance, SBOMs, and Threat Modeling
- 8.Design Controls for Medical Devices: 21 CFR 820.30 in Practice
- 9.AI/ML SaMD: FDA Artificial Intelligence Guidance in Practice
- 10.PCCP for AI SaMD: FDA Predetermined Change Control Plans
- 11.SaMD Engineering Toolchain: How Small Teams Ship FDA-Cleared Software