ISO 14971 Risk Management for SaMD: What FDA Reviewers Read
By Zakaria El Houda
ISO 14971:2019 is the internationally recognized standard for applying risk management to medical devices, including software as a medical device (SaMD). It defines the process for identifying hazards, estimating and evaluating risks, implementing controls, and monitoring residual risk through the product's entire lifecycle. The FDA recognizes it as a consensus standard.
FDA reviewers don't read your 510(k) submission front to back. They skip to the risk management file. For ContourCompanion, an IMDRF Category IV AI SaMD that generates radiation therapy contours, the risk management file was the single document where reviewers spent the most time. If the hazard analysis was sloppy or the traceability chain had gaps, nothing else in the submission mattered.
ISO 14971:2019 is the internationally recognized standard for medical device risk management. The FDA recognizes it as a consensus standard. But reading it cover to cover won't tell you how to build a risk management file that works for software. The standard covers everything from implantable pacemakers to surgical robots to standalone software. The concepts transfer. The implementation for SaMD requires interpretation that the standard leaves to you.
ContourCompanion's risk management file had 47 identified hazards. Here's how we structured the analysis, chose our techniques, and built a traceability chain that FDA reviewers could follow without a guided tour.
The Risk Management File: What Actually Goes In It
ISO 14971 Clause 4.5 requires traceability across the entire risk management process. Not a summary. Not a spreadsheet someone filled in before submission. A traceable chain from identified hazard to risk analysis to risk evaluation to risk control to verification of that control to residual risk evaluation.
The file covers nine clauses of ISO 14971: the risk management plan (Clause 4.4), intended use and foreseeable misuse (5.2), hazard identification (5.3-5.4), risk estimation (5.5), risk evaluation (6), risk controls (7.1), verification of those controls (7.2), overall residual risk (8), and the risk management review (9). You can read the clause requirements on the ISO catalog page. What matters is how they connect.
The sections that consumed the most effort were hazard identification and risk control verification. Identifying dozens of hazards across algorithm, data handling, integration, infrastructure, and operator error took multiple sessions with clinicians, the algorithm team, and the engineering team. Each session surfaced hazards that the others hadn't considered. The radiation oncologists identified operator-side failure modes (wrong time point selected, automation bias) that the engineering team would have missed. The engineering team identified infrastructure failure modes (silent OOM, network timeout without notification) that had non-obvious clinical consequences. Running these sessions with mixed expertise isn't optional.
The FDA submission included a Device Hazard Analysis table as a summary. The full risk management file lived in the Design History File. Reviewers requested the full file during review. It needs to hold up on its own.
Medical Device Risk Management: Setting Criteria Before You Start
The risk management plan must define acceptability criteria before you begin the analysis. This is Clause 4.4, and teams routinely get it backwards: they identify hazards first, then set criteria that conveniently make everything acceptable. Auditors and FDA reviewers recognize this pattern immediately.
We used a 3x3 severity-probability matrix. Severity: Negligible (will not impact clinical care), Moderate (reversible or minor impact to clinical care), Significant (negatively impacts clinical care). Probability: Low (unlikely, rare, remote), Medium (can happen but not frequently), High (likely, often, frequent).

R1, R2, R3, and R6 were unacceptable. The rest were acceptable. No ALARP zone.
There's a deliberate choice here. A 5x5 matrix with an ALARP band is more common in the industry, and it's what ISO/TR 24971 examples show. We went with 3x3 because it forces binary decisions. With a 5x5, teams hide risks in the ALARP zone and defer the hard conversation about whether a risk is actually acceptable. A 3x3 makes you commit: either you accept it or you control it. For a Category IV SaMD where the top hazards involve radiation to unprotected organs, ambiguity in the matrix is worse than a few extra controls.
The probability categories were calibrated to usage estimates: expected product lifespan, number of clinical sites, contours generated per site per day. "Low" meant rare enough that it wouldn't be expected during the product's deployed lifetime at the planned install base. These numbers feed directly into the probability estimation for each hazard, so they need to be defensible.
Hazard Analysis for Software: Not Hardware FMEA
ISO 14971 doesn't prescribe a specific analysis technique. It references Fault Tree Analysis, FMEA, HAZOP, and Preliminary Hazard Analysis in its annexes (now moved to ISO/TR 24971:2020). For SaMD, we used a combination of FTA and FMEA.
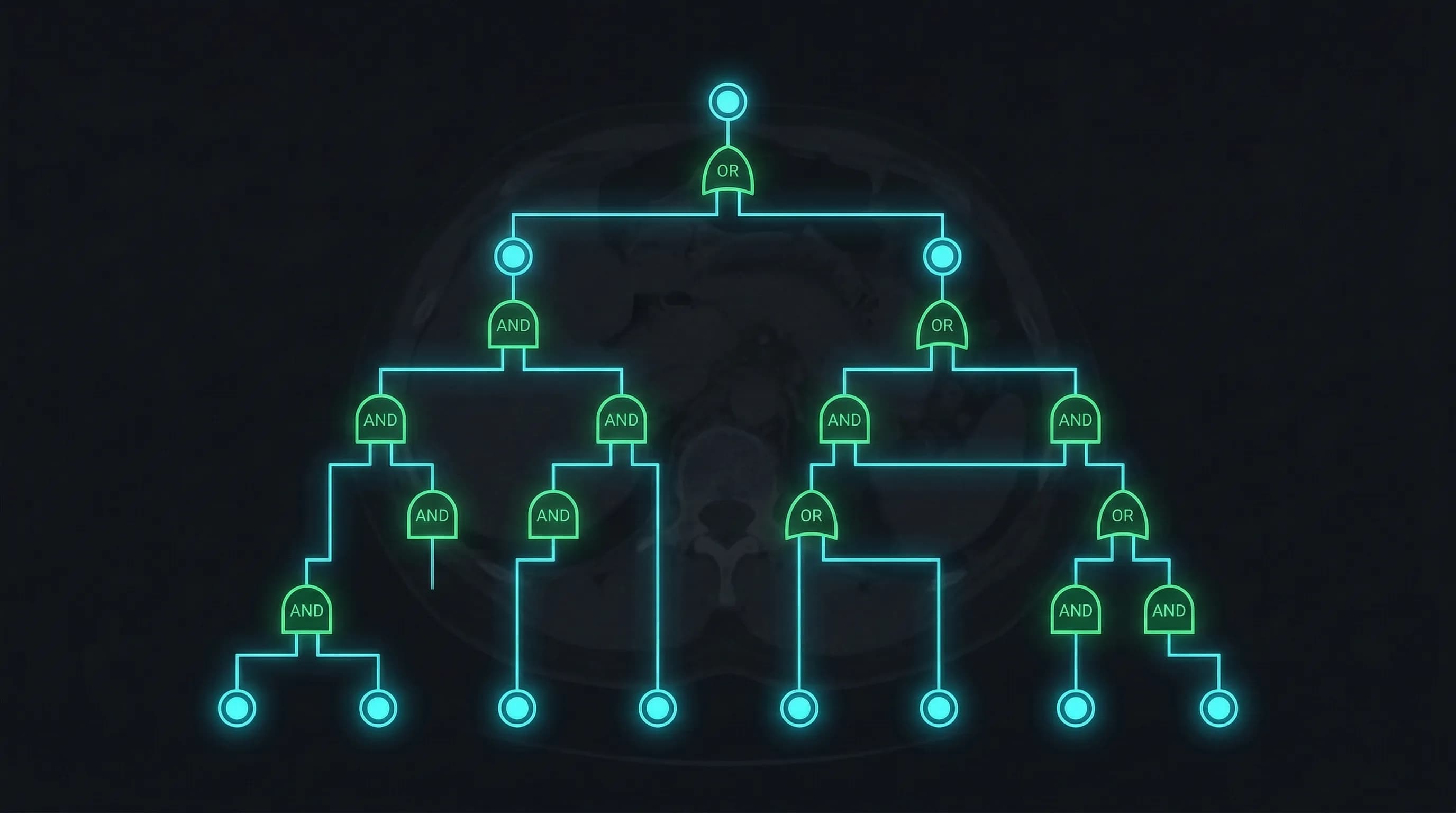
Fault Tree Analysis identified the hazard structure
FTA is top-down: start with the worst outcome and decompose backward through AND/OR gates to find every combination of basic events that could cause it. For ContourCompanion, the top-level event was "Segmented contours are not accurate." That decomposed into seven branches:
- Algorithm Error: wrong parameters, wrong input data, internal error
- Data Error: I/O errors, read/write errors, file handle errors, DICOM format errors
- Operator Error: wrong patient position, wrong time point selected
- Image Quality: insufficient image quality for contouring, motion artifacts
- Display Error: coordinate system error, orientation error, time point error
- Network Error: transmission failures between components
- Positioning Error: patient positioning data inconsistent with imaging data

Each branch decomposed further into basic events. The value of FTA over FMEA alone is that it reveals combinatorial failures: two individually acceptable risks that become unacceptable when they co-occur. A minor data error combined with an operator who doesn't verify the output is a different risk profile than either one alone. FMEA analyzes single failure modes. FTA models the gates between them.
FMEA quantified the individual risks
Once the fault tree established the hazard structure, we used FMEA to estimate severity and probability for each failure mode and assign controls. Software FMEA requires a different mindset than hardware FMEA. Software doesn't wear out or degrade. Software failures are deterministic: they result from design defects, specification gaps, or environmental conditions the design didn't account for.
For each software function, we asked three questions: what can go wrong (failure mode)? What happens to the patient if it goes wrong (effect)? What causes it (root cause)?
A condensed view illustrating how this looks for an autocontouring SaMD:
| Function | Failure Mode | Clinical Effect | Sev | Cause | Prob | Controls |
|---|---|---|---|---|---|---|
| Contour generation | Under-segmented structure | Radiation damages organ at risk | Significant | Model undertrained on morphology variants | Medium | Accuracy threshold; flagged to clinician for review |
| DICOM parsing | Wrong patient data processed | Contours applied to wrong patient | Significant | Frame of reference UID not validated | Low | UID validation against worklist; patient ID cross-check |
| Result delivery | Contour silently dropped by TPS | Treatment planned without OAR protection | Significant | RT Structure Set formatting error | Low | DICOM conformance testing per PS3.3; integration testing against target TPS systems |
| Inference engine | OOM crash during processing | Clinician unaware contour wasn't generated | Moderate | Input volume exceeds memory allocation | Low | Resource bounds testing; timeout with explicit failure notification |
Notice the severity column. An OOM crash is a minor software event. The clinical consequence, the clinician not knowing the contour wasn't generated, is what drives severity. Teams that rate severity based on the software behavior instead of the clinical impact systematically underrate their risks. FDA reviewers catch this.
ISO 14971 Risk Control: The Three-Tier Hierarchy
ISO 14971 Clause 7.1 requires controls in priority order. This isn't a suggestion. Regulators expect justification for why you chose a lower-priority control when a higher-priority one was feasible:
-
Safety by design: eliminate the hazard entirely. We used strongly typed interfaces to prevent invalid DICOM data from reaching the inference engine, and built fail-safe behavior so the system refuses to produce output rather than producing potentially wrong output.
-
Protective measures: reduce risk through safeguards you add on top of the design. For an autocontouring SaMD, this includes accuracy thresholds that flag uncertain cases to the clinician, input validation that rejects incomplete or malformed imaging data, automated DICOM conformance checking, and resource bounds enforcement with explicit timeout behavior.
-
Information for safety: warnings and instructions when design and protective measures can't fully eliminate the risk. On-screen alerts when results fall below quality thresholds, IFU documenting the intended patient population and imaging protocol requirements, and visual indicators highlighting areas that need closer clinical review.
Each risk control became a software requirement in the IEC 62304 lifecycle. The traceability chain runs: hazard (risk management file) to risk control (risk management file) to software requirement (SRS) to implementation (PR) to verification (test result). That chain is what FDA reviewers follow. If any link is missing, they issue an Additional Information request.

As covered in Post 3, this traceability was enforced by tooling, not discipline. Requirements carried unique IDs. Test cases referenced the requirement they verified. PRs referenced the requirement they implemented. The traceability matrix was generated from these links, not maintained by hand.
Verifying Risk Controls: Proving They Work
Identifying a control isn't enough. ISO 14971 Clause 7.2 requires verification that each control is implemented correctly, reduces risk as intended, and doesn't introduce new hazards. The verification approach for software risk controls, functional tests, boundary tests, fault injection, and negative tests, is covered in detail in the IEC 62304 unit verification section of this series.
The piece specific to risk management: every test that verifies a risk control must reference the hazard ID, control ID, and requirement ID in the test report. When an FDA reviewer asks "How do you know risk control RC-017 works?", the answer is a test report with a pass/fail result, not a statement of intent. We generated this evidence automatically through the CI pipeline, so every build produced a current verification record. Teams that defer risk control verification to a pre-submission manual pass are betting that nothing broke since the last time they checked.
Overall Residual Risk: The 2019 Requirement Teams Miss
ISO 14971:2019 added Clause 8 as a dedicated requirement: evaluate the overall residual risk profile, not just the residual risk from each individual hazard. Dozens of individually acceptable residual risks can combine to create an unacceptable risk profile. A system with ten controlled hazards in the same failure pathway has a different aggregate exposure than one with ten controlled hazards spread across independent subsystems.
For ContourCompanion, the overall evaluation considered cumulative exposure: hundreds of contours per clinical site per week, across multiple anatomical structures, each feeding directly into a treatment planning system. Even with individually low probabilities, the aggregate volume creates a meaningful expected frequency over the product's installed base.
If overall residual risk is unacceptable and cannot be further reduced, the standard requires a benefit-risk analysis. For ContourCompanion, we structured it as a quantitative comparison: manual contouring error rates from published literature versus the controlled residual risk of automated contouring. Published studies show AI autocontouring reduces contouring time by 69% with mean DSC of 0.81-0.86, and manual contouring has its own well-documented inter-observer variability. The argument wasn't that ContourCompanion is risk-free. It was that the residual risk with all controls in place is lower than the baseline risk of the manual process it replaces, and the time savings directly benefit patient throughput.
Post-Production: The Risk File Is a Living Document
ISO 14971:2019 expanded Clause 10 significantly from the 2007 edition. Post-production information collection is no longer passive. The standard requires active monitoring for new hazards, changes in probability estimates, and signals that the original risk analysis is no longer valid.
For a cloud-deployed SaMD, post-production monitoring pulls from the product itself (processing success/failure rates, quality metric distributions, performance telemetry), service records, user feedback, outcomes research from clinical sites, and changes in applicable regulations and standards. Quality metric distributions are the most useful signal: if the percentage of below-threshold results increases for a specific use case, that is an early indicator that the production population may differ from the validation dataset. That signal triggers a risk reassessment before it becomes a complaint.
The risk management file updates through the CAPA process. New hazards trigger a risk assessment, new controls go through verification, and the overall residual risk evaluation gets updated. Management reviews the process annually. Post-market surveillance and CAPA deserve their own post in this series. The structural point: the risk management file stays open for the life of the product.
Common Deficiencies That Trigger Additional Information Requests
Based on our experience and published audit findings:
- Missing benefit-risk analysis. If any residual risk sits in a zone that requires justification, the file needs a benefit-risk argument. Teams leave this out more than any other element.
- Traceability that stops halfway. Hazards identified but not traced to controls, or controls identified but never linked to verification evidence. The chain has to run unbroken from hazard to test result. One missing link and the reviewer sends an AI request.
- Criteria set after the analysis. The plan must define acceptability criteria before hazard identification begins. Criteria that conveniently make all risks acceptable are a red flag.
- Skipping Clause 8 entirely. Teams evaluate individual risks and never step back to ask whether the aggregate residual risk profile is acceptable. The 2019 edition made this an explicit, separate requirement, and auditors check for it.
- Post-production plan missing or vague. "We will monitor complaints" is not a plan. The plan specifies what information you'll collect, how you'll review it, what triggers a reassessment, and how often management reviews the process.
- Rating severity on software behavior instead of clinical consequence. We covered this above with the OOM example. It's the most common mistake in software FMEA and the easiest one for FDA reviewers to spot.
Frequently Asked Questions
What is the difference between ISO 14971:2019 and ISO 14971:2007?
The 2019 edition added Clause 8 as a dedicated requirement for overall residual risk evaluation, not just individual hazard assessment. It also expanded Clause 10 to require active post-production monitoring, not passive complaint collection. The acceptability criteria structure and risk control hierarchy remained the same, but the 2019 edition is more explicit about benefit-risk analysis and post-market obligations. If you're starting a new project, build to the 2019 edition.
Should I use a 3x3 or 5x5 risk matrix for SaMD?
Either can satisfy ISO 14971. We used a 3x3 matrix for ContourCompanion because it forces binary accept/reject decisions. A 5x5 matrix with an ALARP (As Low As Reasonably Practicable) band gives teams a place to park ambiguous risks without committing to whether they're acceptable. For a Category IV SaMD where top hazards involve radiation to unprotected organs, that ambiguity is worse than implementing a few extra controls. The matrix dimensions matter less than whether your probability categories are calibrated to real usage estimates.
What is the difference between FTA and FMEA for medical device software?
Fault Tree Analysis (FTA) is top-down: start with the worst outcome and decompose backward to find every combination of events that could cause it. FMEA is bottom-up: start with individual failure modes and assess their effects. FTA reveals combinatorial failures where two individually acceptable risks become unacceptable together. FMEA quantifies severity and probability for individual failure modes. For SaMD, using both together gives the most complete picture. FTA establishes the hazard structure, FMEA fills in the risk estimates and controls.
How many hazards should a SaMD risk management file identify?
There's no fixed number. ContourCompanion's file identified dozens of hazards across algorithm, data handling, integration, infrastructure, and operator error categories. The number depends on your intended use, clinical context, and system architecture. A simple Class A SaMD might have 10-15 hazards. A Class C AI SaMD that integrates with treatment planning systems will have more. The test isn't the count: it's whether you can justify that your identification process was thorough enough that a reasonable hazard wouldn't be missing.
References
- ISO 14971:2019 — Risk Management for Medical Devices
- ISO/TR 24971:2020 — Guidance on the Application of ISO 14971
- IEC 62304:2006+AMD1:2015 — Medical Device Software Lifecycle Processes
- 21 CFR Part 820 — Quality System Regulation
- FDA: Content of Premarket Submissions for Device Software Functions (June 2023)
- AAMI TIR57:2016 — Principles for Medical Device Security Risk Management
- Real-world autocontouring validation (BJR 2024)
SaMD Engineering
Part 4 of 11
- 1.What Is SaMD? Software as a Medical Device Explained
- 2.FDA Pathways for SaMD: 510(k) vs De Novo vs PMA
- 3.IEC 62304 in Practice: Medical Device Software Without the Waterfall
- 4.ISO 14971 Risk Management for SaMD: What FDA Reviewers Read
- 5.HIPAA Compliant AWS Architecture for Medical Device Software
- 6.Infrastructure as Code for Medical Devices: IQ OQ PQ with AWS CDK
- 7.Medical Device Cybersecurity: FDA Guidance, SBOMs, and Threat Modeling
- 8.Design Controls for Medical Devices: 21 CFR 820.30 in Practice
- 9.AI/ML SaMD: FDA Artificial Intelligence Guidance in Practice
- 10.PCCP for AI SaMD: FDA Predetermined Change Control Plans
- 11.SaMD Engineering Toolchain: How Small Teams Ship FDA-Cleared Software